علائم و درمان بیماری تالاسمی مینور و ماژور چیست

دلایل معده درد که به آن بی توجهیم
دلایل معده درد که به آن بی توجهیم معده درد,بيماري معده درد,معده درد در بارداري,علائم معده درد,درمان معده درد,درمان معده درد با طب سوزني,مواد غذايي مناسب براي درمان معده درد,معده درد را …

راه های درمان التهاب بینی
راه های درمان التهاب بینی التهاب بینی,بيماري التهاب بینی,التهاب بینی در بارداري,علائم التهاب بینی,درمان التهاب بینی,درمان التهاب بینی با طب سوزني,مواد غذايي مناسب براي درمان التهاب بینی,التهاب بینی را چگونه درمان کنيم,التهاب …
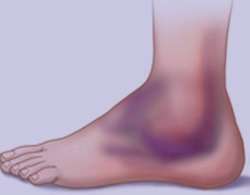
کوبيدگی و خونمُردگی در عضلات
کوبيدگی و خونمُردگی در عضلات کوبيدگی و خونمُردگی در عضلات,بيماري کوبيدگی و خونمُردگی در عضلات,کوبيدگی و خونمُردگی در عضلات در بارداري,علائم کوبيدگی و خونمُردگی در عضلات,درمان کوبيدگی و خونمُردگی در عضلات,درمان کوبيدگی …
تالاسمی
تالاسمی ,تالاسمی مینور,تالاسمی ماژور,تالاسمی آلفا,تالاسمی مینور و ازدواج,تالاسمی چیست,تالاسمی مینور ازدواج,تالاسمی مینور چیست؟,تالاسمی ماژور چیست,تالاسمی مینور و عوارض آن,تالاسمی مینور و بارداری,تالاسمی مینور درمان,تالاسمی مینور در کودکان,تالاسمی مینور در بارداری,تالاسمی مینور و ماژور,تالاسمی ماژور,تالاسمی ماژور و ازدواج

تالاسمی
تالاسمی از بیماریهای ژنتیکی است که در اثر آن هموگلوبین ساختار طبیعی خود را از دست میدهد وبنابراین پدیدهٔ تولید هموگلوبین غیر مؤثر در بدن ایجاد میشود در نتیجه هموگلو بین معیوب قادر به اکسیژن رسانی مطلوب به اعضا بدن نیست. پس در واقع کمبود کلی هموگلوبین وجود ندارد، بلکه هموگلوبین غیرطبیعی افزایش یافتهاست. هموگلوبین جزء انتقال دهنده اکسیژن در سلولهای قرمز خونی است. هموگلوبین شامل دو پروتئین مختلف به نام آلفا و بتا است. اگر بدن توانایی تولید کافی از هر نوع پروتئین را نداشته باشد، سلولهای خونی بطور کامل شکل نمیگیرند و توانایی انتقال اکسیژن کافی را ندارند و نتیجه یک نوع کم خونی است که در طفولیت آغاز میشود و تا پایان عمر به طول میانجامد. هر چند این بیماری یک اختلال منفرد نیست، اما یک گروه اختلالات از طرق مشابه بدن انسان را درگیر میکند. درک تفاوت بین گونههای مختلف این بیماری مهم است.
تالاسمی مینور
اشخاص با خصیصه تالاسمی مینور دارای این بیماری هستند ولی بیمار نیستند این اشخاص مطلقا” سالم و طبیعی هستند ، بعضی از آنها ممکن است کم خونی خفیف داشته باشند بیشتر کسانی که حامل خصیصه این بیماری هستند نمی دانند که ناقل تالاسمی مینور هستند فقط وقتی از آن مطلع می شوند که خونشان را آزمایش کنند و یا صاحب یک کودک مبتلا به تالاسمی ماژور شوند . گلبولهای قرمز خون اشخاصی که تالاسمی مینور دارند کوچکتر حد معمول است . در خون مبتلایان به خصیصه تالاسمی مینور همچنین از یک نوع هموگلوبین که هموگلوبین A2 نامیده می شوند مختصری بیشتر از حد معمول وجود دارد . خصیصه تالاسمی مینور د ر موقع تولد وجود دارد و در تمام طول زندگی باقی می ماند.ممکن است توسط والدین به کودکان منتقل شود یعنی ارثی است .دانستن اینکه تالاسمی مینور دارید مهم است زیرا گاهی ممکن است از اشخاصی که خصیصه تالاسمی مینور دارند صاحب فرزند مبتلا به تالاسمی ماژور شوید . که اختلال خونی شدیدی است.
تالاسمی ماژور
تالاسمی ماژور یا آنمی کولی به علت حذف یا جهش در هر دو ژن سازنده زنجیره بتا ایجاد می شود و به این ترتیب یا هیچ زنجیره بتایی ساخته نمی شود و یا به مقدار کمی ساخته می شود. در نتیجه بدن کمبود این زنجیره ها را با ساخت زنجیره های آلفا جبران می کند که این زنجیره های آلفای اضافی برای گلبولهای قرمز سمی هستند و با رسوب بر روی سلولهای گلبول قرمز باعث می شوند که گلبولهای قرمز در مغز استخوان و در داخل خون تخریب شده و زنجیره های آلفا در مغز استخوان رسوب می کنند. از طرفی به علت خونسازی غیر موثر ، مراکز خونساز خارج مغز استخوان ، ار جمله کبد و طحال شروع به خونسازی می کنند و بزرگ می شوند. بیماری معمولا بصورت کم خونی شدید در 6 ماهه اول زندگی کودک تظاهر می کند و درصورت عدم شروع تزریق خون ، بافت مغز استخوان و مکانهای خونساز خارج مغز استخوان فعال و بزرگ شده و باعث بزرگی مغز استخوانها بخصوص استخوانهای پهن (صورت و جمجمه) و بزرگی کبد و طحال می شوند.
تالاسمی چیست
این بیماری نوعی کم خونی ارثی و ژنتیکی است که به علت اشکال در ساخت زنجیره های پروتئینی هموگلوبین (ماده اصلی داخل سلولهای قرمز خون) ایجاد می شود. هموگلوبین نه تنها برای حمل و تحویل طبیعی اکسیژن لازم است، بلکه در شکل و اندازه و بدشکلی گلبول قرمز نیز دخالت دارد.تالاسمی کودکان هموگلوبین مولکول اصلی داخل گویچه های قرمز است که از هم و زنجیره های پروتئنی یا گلوبین تشکیل شده است. در هر زنجیره گلوبین یک مولکول هم وجود دارد که اکسیژن را توسط آهن خود حمل می کند. پس تولید هموگلوبین نیاز به تامین آهن و ساخت هموگلوبین دارد بر اساس نوع زنجیره پروتئینی چند نوع هموگلوبین وجود دارد.
تالاسمی آلفا
به گزارش سایت جسارت انتقال اکسیژن در خون توسط گلبولهای قرمز و پروتئینی به نام هموگلوبین انجام میگیرد . این پروتئین از دو زنجیره آلفا و دو زنجیره بتا تشکیل شده است. کمبود هر کدام از این زنجیرهها که در اثر نقصان عملکرد ژنهای تولید کننده آنها بروز میکند باعث بروز بیماری کم خونی ژنتیکی موسوم به تالاسمی میشود. تالاسمی آلفا به علت کاهش میزان زنجیره آلفا گلوبولین ایجاد میشود. این بیماری به صورت اتوزومال مغلوب به ارث میرسد. پیش از انجام آزمایش ژنتیکی لازم است آزمایشات CBC و الکتروفورز هموگلوبین انجام گیرد. تشخیص ژنتیکی این بیماری در دو مرحله تشخیص ناقلین (مرحله اول) و تشخیص پیش از تولد (مرحله دوم ) انجام میگیرد. با انجام آزمایش مرحله اول در صورت نیاز بر اساس نوع ژنوتیپ والدین ضرورت بررسی مرحله دوم برای جنین مشخص میشود. کلاستر ژن آلفا گلوبولین در انتهای بازوی کوتاه کروموزم 16 قرار دارد. در داخل این کلاستر دو ژن آلفا یک و آلفا دو قرار دارند که تولید کننده زنجیره آلفا هستند. همچنین یک ژن کاذب و یا ژن غیر فعال آلفا نیز در این کلاستر وجود دارد. ژن زتا (ζ) که تولید کننده زنجیره آلفا در دوران جنینی (embryo) است به همراه پسودوژن زتا نیز در این کلاستر قرار دارند. به دلیل وجود تشابه 95 درصدی ژنهای آلفا یک و آلفا دو احتمال جفت شدن نابجای این دو توالی و در نتیجه حذف ژنی وجود دارد. حذف بخشی از ژن آلفا گلوبولین در بیش از 90 درصد موارد عامل ایجاد کننده بیماری است. در 10 درصد موارد نیز حذفهای کوچک باعث به وجود آمدن علائم بیماری می شوند. افراد سالم 4 نسخه ژن فعال آلفا (دو تا بر روی هر کروموزم 16) دارند. فردی که یک نسخه ژن غیر فعال و 3 نسخه ژنی فعال داشته باشد به عنوان ناقل خاموش آلفا تالاسمی شناخته میشود (α α /− α). تاثیر نقصان یک زنجیره آلفا در ناقلین خاموش به حدی کم است که تاثیری در عملکرد پروتئین هموگلوبین و باتبع سلامت فرد حامل ندارد. افرادی که دو نسخه فعال و دو نسخه غیر فعال داشته باشند ناقلان دارای خصیصه آلفا تالاسمی نامیده میشوند. این افراد دچار کمخونی خفیف هستند که نشانه بالینی ندارد و معمولا با فقر آهن اشتباه گرفته میشود. البته نحوه توزیع ژنهای معیوب آلفا تالاسمی بر روی دو کروموزم و یا ژنوتیپ ناقلان دارای خصیصه آلفا تالاسمی به صورت سیس (α α /− −) و یا ترانس (α −/ α −) است. دو نوع مهم آلفا تالاسمی که با عوارض متعدد بالینی رو به رو هستند آلفا تالاسمی ماژور یا هیدروپس فتالیس با ژنوتیپ (− − /− −) و بیماری H با ژنوتیپ (− −/− α) است. مبتلایان به بیماری H در بدو تولد با علائمی چون کوچک بودن گلبولهای قرمز (microcytic)، کمخونی همولیتیک هایپرکرومیک (hypochromic hemolytic anemia)، بزرگی کبد و طحال (hepatosplenomegaly)، زردی و در مواردی تغییرات استخوانی مرتبط با تالاسمی روبهرو هستند. این بیماران نیاز به تزریق خون دارند. در جنینهای مبتلا به هیدروپس فتالیس یا تالاسمی آلفا ماژور نیز نوع خاصی هموگلوبین که از چهار زنجیره لاندا تشکیل شده و هموگلوبین Barts نامیده میشود در خون تجمع مییابد. این جنینهای مبتلا معمولا پیش از تولد یا مدت کوتاهی پس از تولد فوت میکنند. در این موارد علاوه بر خود جنین، جان مادران جنینها نیز در طول دوران بارداری و یا در زمان وضع حمل در معرض خطر است. زمانی که هر دو والد ناقل دارای خصیصه آلفا تالاسمی به صورت ترانس باشند، هیچ گاه فرزند بیمار به دنیا نمیآید و تمام فرزندان مشابه والدین ناقل این بیماری دارای خصیصه این بیماری خواهند بود.
تالاسمی ازدواج
سید حسینعلی صابری در ارتباط با موضوع این بیماری اظهارداشت : تشخیصهای انجام شده قبل از تولد به صورت ۱۰۰ درصد و تضمینی مانع از تولد کودک ناهنجار میشود اما با توجه به هزینه های بالای ژنتیک در جهان و ایران، از ازدواج دو فرد مبتلا به نوع مینور جلوگیری می شود. پزشک مشاور ژنتیک با اشاره به اینکه نوع ماژور حاصل ژنهای ناقل و معیوبی است که بیشتر در اثر ازدواج های فامیلی صورت میگیرد عنوان کرد: همچنین این بیماری در بعضی از نقاط مثل شمال کشور ، استانهای گیلان و مازندران و جنوب مثل هرمزگان و بوشهر شیوع بالاتری دارد و این مناطق بر روی خط تالاسمی قرار دارند. صابری افزود: در این مناطق حتی در ازدواج های غیرفامیلی به دلیل اینکه ناقلیت ژن معیوب تالاسمی در بین مردم شیوع فراوانی دارد احتمال بروز این بیماری نیز نسبت به دیگر مناطق کشور بیشتر است و در نتیجه به طور اتوماتیک دانشگاههای علوم پزشکی و مراکز بهداشت این افراد را مورد ارزیابی قرار میدهند. وی ضمن بیان اینکه تالاسمی ماژور یا کشنده با کم خونی بروز میکند، افزود: از علائم دیگر این بیماری به غیر از کم خونی ، بد شکلی های ظاهری در سروصورت و اندامها و بزرگی کبد و طحال است. بیماران مبتلا به نوع ماژور به مرور زمان با استفاده از جراحی طحال را از بدن خارج می کنند و حتی از شش ماهگی به بعد نیاز به تزریق خون خواهند داشت. پزشک مشاور ژنتیک با بیان اینکه غربالگری قبل از ازدواج برای جلوگیری از شیوع این بیماری خطرناک ضروری است، توضیح داد: چنانچه خانم یا آقا به کم خونی مبتلا باشند باید قبل از ازدواج مورد بررسی و معالجه قرار بگیرد. صابری، تأکید کرد: خوشبختانه ایران به عنوان یکی از موفقترین کشورها در دنیا در رابطه با این بیماری و غربالگری قبل از ازدواج تالاسمی شناخته شده است و بر این اساس باید متذکر شد امروزه ناقلیت این بیماری عاملی برای ممنوعیت ازدواج در جهان نیست ولی در ایران با توجه به عدم دسترسی به آزمایشات ژنتیک و عدم ارزیابی اجباری دولتی و منظم در طول بارداری، از این ازدواج ها جلوگیری می شود که امری منطقی برای شرایط فعلی کشور ما و بسیاری کشورهای منطقه است.
تالاسمی مینور چه بخورند
ویتامین C عامل افزایش دهنده ی جذب آهن است. ویتامینC در میوه ها (به ویژه در مرکبات و گوجه فرنگی) و سبزی ها (جعفری، شاهی، شنبلیله، تره، شبت، گل کلم، فلفل دلمه ای و…) یافت می شود. سبزی ها ی پخته شده حاوی ویتامین C کمتری می باشند. برای مثال مصرف یک عدد پرتقال یا 100 گرم سبزی خوردن همراه با غذا جذب آهن را دو برابر خواهد کرد. بنابراین بیماران تالاسمی عزیز بهتر است از مصرف میوه ها و سبزی ها به همراه وعده ی غذایی یا بلافاصله پس از آن خودداری کنند. اما از آنجایی که میوه ها و سبزی ها، حاوی انواع ویتامین ها و آنتی اکسیدان ها می باشند و مصرف آن ها ضروری است، بهتر است در بین دو وعده ی اصلی غذا (به عنوان میان وعده) مصرف شوند. * گوشت قرمز، گوشت پرندگان، ماهی و سایر غذاهایی دریایی این گروه از مواد غذایی نه تنها حاوی مقدار زیادی آهن از نوع هِم می باشند، بلکه باعث افزایش جذب آهن غیرهِم از سایر غذاها نیز می شوند.
تالاسمی مینور و بارداری
اگر شما به این بیماری مبتلا باشید، متخصص زنان و زایمان شما را به یک متخصص اختلالات خونی (هموتولوژیست) ارجاع میدهد تا وی به شما کمک کند. بسته به نوع این بیماری که به آن مبتلا هستید، شما در دوران بارداری تحت نظارت یک تیم متخصص قرار میگیرید. چه شما به این بیماری مبتلا باشد و چه حامل آن باشید، باید کودک شما 5 میلیگرم اسید فولیک در روز در دوران بارداری دریافت کند زیرا ابتلا به این بیماری احتمال این که لولههای عصبی کودک مانند اسپینا بیفیدا (مهره شکافدار) معیوب باشد را افزایش مییابد. مصرف مقادیر زیاد فولیک اسید در روز احتمال ابتلا به این عوارض را کاهش میدهد. مصرف روزانه فولیک اسید باعث میشود که خون شما سالمتر بماند. ابتلا به این بیماری منجر به کمخونی در دوران بارداری میشود. اگر شما به نوع بتای مینور مبتلا باشید، پزشک قبل از تجویز قرص آهن دستور انجام آزمایشهای تکمیلی برای مشخص کردن میزان سطح آهن را میدهد. نوع بتا مینور بر نتایج آزمایش خون در دوران بارداری اثر داشته و ممکن است نشاندهنده پایین بودن سطح آهن خون باشد در حالی که در واقعیت این گونه نیست. نوع آلفای خفیف میتواند باعث ابتلا به کمخونی شود به خصوص اگر شما حامل دو ژن باشید. نوع آلفا HbH ممکن است باعث کمخونی خفیف تا شدید شده و باعث شود که شما نیاز به تزریق خون در دوران بارداری پیدا کنید. تالاسمی بتای ماژور احتمال ابتلا به عوارض را در دوران بارداری افزایش میدهد. ارگانهای شما که تحت فشار هستند برای رشد نوزاد نیاز و تقاضاهای بیشتری دارند. انتقال خون و نیاز به مصرف برخی داروها ممکن است روند بارداری شما را تغییر دهد.
تالاسمی مینور در کودکان
این بیماری به دو نوع آلفا و بتا است که خود بتا هم شامل ماژور (شدید) و مینور یا خفیف می باشد. افراد مبتلا به مینور در واقع کم خونی مشکل سازی ندارندریا، ولی اگر دو فرد مینور با هم ازدواج کنند به احتمال 25% فرزندنشان دچار نوع شدید ماژور خواهد بود و 25% فرزندشان سالم و 50% نوع مینور خواهند داشت.
بیماری تالاسمی
این بیماری به گروهی از اختلالات ژنتیکی در خون اطلاق می شود. برای فهم تأثیر این بیماری بر بدن انسان لازم است ابتدا درباره نحوه ساخته شدن خون در بدن نکاتی را بدانیم. هموگلوبین جزو انتقال دهنده اکسیژن در سلول های قرمز خونی است. هموگلوبین شامل ۲ پروتئین مختلف به نام آلفا و بتا است. اگر بدن توانایی تولید کافی از هر نوع پروتئین را نداشته باشد، سلول های خونی به طور کامل شکل نمی گیرد و در این صورت توانایی انتقال اکسیژن کافی را ندارد و در نتیجه نوعی کم خونی در بدن ایجاد می شود که شاید تا پایان عمر همراه فرد باشد. این بیماری که یکی از شایع ترین بیماری ها در ایران است معمولا از پدر و مادری که ناقل ژن کم خونی است به فرزندان به ارث می رسد. گاهی کم خونی و فقرآهن با تالاسمی اشتباه گرفته می شود. اولین قدم برای تشخیص این بیماری تعیین موتاسیون است. باید بررسی شود که پدر و مادر چه نوع موتاسیونی دارند. به طور مثال، کم خونی داسی شکل ۳۰۰ یا ۴۰۰ موتاسیون دارد.
درمان تالاسمی مینور چیست
کم خونی یک اصلاح کلی و عامیانه است و انواع مختلفی دارد. شایع ترین آن کم خونی فقر آهن و تالاسمی است. باید مشخص شود که کم خونی فرزند شما از چه نوع است. شما حتما باید یک آزمایش کم خونی برای فرزند خود بدهید تا مشخص شود نوع کم خونی او مینور ،ماژور و یا از نوع دیگری است. با توجه به نوع آن نیز نوع درمان مشخص می شود. موارد این بیماری نیز ژنتیکی است که از پدر و مادر می رسد. اگر مشخص شود که نوع کم خونی فرزند شما تالاسمی است نباید درمان را قطع کنید. شما باید اسیدفولیک و آهن را به طور همزمان مصرف کنید که میزان آن نیز توسط دکتر و بر اساس نوع و شدت آن مشخص می شود. ممکن است به طور مداوم این مشکل در آزمایش خون فرزندتان وجود داشته باشد و باید به طور مداوم داروهایش را مصرف کند. این مشکل درمان ریشه کن ندارد ولی تا زمانی که اسیدفولیک را مصرف کند آزمایش خون او نیز نرمال گزارش می شود. اما فقر آهن اینجوری نیست. برای درمان اگر ذخایر استخوان بچه از آهن پر شود دیگر نیازی به ادامه درمان نیست. نوع ماژور عوارض زیادی دارد که در مورد فرزند شما نیست و درمان آن باید با تزریق خون انجام شود. نوع مینور اما عارضه خاصی ندارد. نگران نباشید.
علائم تالاسمی مینور
ا – رنگ پریدگی زبان و مخاط داخل لب و پلک چشم 2-خستگی زود رس 3-بی تفاوتی 4- سرگیجه و سردرد 5- خواب رفتن و سوزن سوزن شدن دست و پا 6 – حالت تهوع 7 – در کم خونی های شدید گود شدن روی ناخن
انواع تالاسمی
دو نوع تالاسمی داریم آلفا و بتا که آلفا به چهار قسمت و بتا به قسمت تقسیم میشوند
آلفا :حامل خاموش – نوع Trait یا خفیف و ملایم – هموگلوبین H – هیدروپس فتالین یا آلفا تالاسمی ماژور
بتا :نوع مینور یا نوع Trait – نوع اینترمدیا یا متوسط – نوع ماژور یا آنمی کولی
عوارض تالاسمی
از عوارض این بیماری یک سری از عوارض از جمله اختلال رشد و تغییر قیافه بیمار و بزرگی کبد و طحال که به علت خود بیماری است و یکسری عوارض دیگر ناشی از درمان این بیماری است. اصلی ترین این عارضه هموسیدوز یا هموکروماتوز است. هموسیدروز به رسوب آهن در بافتها گفته می شود که نتیجه غیر قابل اجتناب تزریق طولانی مدت خون است. در هم نیم لیتر خونی که به بیمار تزریق می شود، حدود 200mg آهن به بافتها منتقل می کند که این مقدار آهن نمی تواند از بدن دفع شود و در بافتها رسوب می کند و باعث نارسایی در بافتها می گردد. بخصوص رسوب آهن در قلب و پانکراس و غدد ، مشکل اصلی این بیماران خواهد بود که باعث نارسایی پانکراس و دیابت ، نارسایی قلبی و نارسایی غدد جنسی و تیروئید و … می شود.
























































هموگلوبین جزو انتقال دهنده اکسیژن در سلول های قرمز خونی است. هموگلوبین شامل ۲ پروتئین مختلف به نام آلفا و بتا است. اگر بدن توانایی تولید کافی از هر نوع پروتئین را نداشته باشد، سلول های خونی به طور کامل شکل نمی گیرد و در این صورت توانایی انتقال اکسیژن کافی را ندارد و در نتیجه نوعی کم خونی در بدن ایجاد می شود که شاید تا پایان عمر همراه فرد باشد.
تالاسمی که یکی از شایع ترین بیماری ها در ایران است معمولا از پدر و مادری که ناقل ژن کم خونی است به فرزندان به ارث می رسد.
گاهی کم خونی و فقرآهن با تالاسمی اشتباه گرفته می شود. اولین قدم برای تشخیص این بیماری تعیین موتاسیون است. باید بررسی شود که پدر و مادر چه نوع موتاسیونی دارند. به طور مثال، کم خونی داسی شکل ۳۰۰ یا ۴۰۰ موتاسیون دارد.
در مورد همسرانی که تالاسمی دارند، هیچ ممنوعیت ازدواج وجود ندارد؛ همسران تحت آزمایش لازم قرار می گیرند و به آنها اجازه داده می شود، در دوره بارداری آزمایش پیش از تولد را انجام دهند. چنانچه این آزمایش انجام نشود، ۲۵ درصد امکان دارد که فرزندشان به تالاسمی مبتلا شود.
راه های پیشگیری از تالاسمی چیست؟
ـ انجام آزمایش خون از نظر کم خونی
ـ انجام آزمایش روی جنین در هفته های اول حاملگی در دوران بارداری
مهم ترین راه درمان تالاسمی، تزریق مداوم خون است. شایع ترین درمان برای تمامی شکل های تالاسمی، تزریق سلول های قرمز خونی است. در صورتی که بیمار در گذشته، به اندازه کافی خون دریافت نکرده باشد، لازم است برای بهبود کیفیت زندگی وی، تعداد دفعات تزریق خون را افزایش دهد.
تالاسمی یک بیماری ارثی خونی می باشد که در آن، بدن میزان هموگلوبین کمتر و یا هموگلوبین های غیر طبیعی تولید می کند. هموگلوبین در اکسیژن رسانی به بخش های مختلف بدن به گلبول های قرمز خون کمک می کند. کم بودن سطح هموگلوبین خون ممکن است منجر به بیماری کم خونی شود. بیماری کم خونی بیماری ای است که باعث می شود شخص مبتلا احساس ضعف و خستگی کند. کم خونی شدید می تواند به اعضای بدن صدمه زده و در نهایت منجر به مرگ فرد مبتلا شود.
تالاسمی دو نوع اصلی دارد: آلفا و بتا که تالاسمی بتا نسبت به تالاسمی آلفا بسیار شایعتر است.
بدن انسان برای تولید هموگلوبین به گلوبین های آلفا و بتا نیاز دارد. تالاسمی نوع بتا زمانی رخ می دهد که یک یا هر دو ژن مولد بتا گلوبین کار نکنند و یا تنها بخشی از وظایفی که به عهده دارند را انجام دهند.
اگر شما فقط یک ژن آسیب دیده داشته باشید ممکن است کم خونی شما خفیف باشد و احتمالا نیازی به درمان نداشته باشد. به این نوع تالاسمی “بتا تالاسمی مینور” گفته می شود. این بیماری زمانی رخ می دهد که شما یک ژن سالم را از یکی از والدین و ژن تالاسمی را از دیگری گرفته باشید.
زمانی که هر دو ژن آسیب دیده باشند به این معناست که شما ژن بیماری تالاسمی را از هر دو والدین دریافت کرده اید. در این حالت ممکن است شما به کم خونی متوسط و یا شدید مبتلا شوید.
اگر شما به کم خونی متوسط (بتا تالاسمی اینترمدیا – beta thalassemia intermedia) دچار شده باشید، ممکن است فرایند انتقال خون لازم شود.
اما اگر به کم خونی شدید (بتا تالاسمی ماژور و یا آنمی کولی – beta thalassemia major or Cooley’s anemia) مبتلا شده باشید، لازم است که در تمام طول زندگی انتقال خون انجام دهید. علائم این نوع کم خونی معمولا در عرض چند ماه پس از تولد بروز می کنند.
این نوع از تالاسمی زمانی رخ می دهد که یک تا چهار ژن آلفا گلوبین آسیب دیده باشند و یا اصلا وجود نداشته باشند.
اگر یکی از ژن ها آسیب دیده و یا وجود نداشته باشد: ممکن است سلول های قرمز خون نسبت به اندازه ی طبیعی خود کوچکتر باشند. این نوع از تالاسمی علائم خاصی نداشته و نیازی به درمان نیز ندارد. اما در این حالت شما یک منتقل کننده ی ژن هستید، به این معنا که می توانید این ژن را به فرزندان خود منتقل کنید.
اگر دو عدد از ژن ها آسیب دیده و یا وجود نداشته باشند: شما به کم خونی بسیار خفیف دچار خواهید شد که معمولا نیازی به درمان ندارد. این نوع از تالاسمی را آلفا تالاسمی مینور (alpha thalassemia minor) می نامند.
اگر سه عدد از ژن ها آسیب دیده و یا وجود نداشته باشند: کم خونی شما متوسط و یا متوسط به بالا خواهد بود. به این نوع از تالاسمی “هموگلوبین H” نیز گفته می شود. ممکن است در حالت های شدید تر آن انجام انتقال خون ضروری شود.
اگر چهار عدد از ژن ها آسیب دیده و یا وجود نداشته باشند: به این نوع از تالاسمی، آلفا تالاسمی ماژور و یا هیدروپس جنینی (hydrops fetalis) گفته می شود. در این بیماری جنین یا مرده به دنیا می آید و یا بعد از تولد خواهد مرد.
نقص در یک یا چند ژن می تواند باعث ایجاد تالاسمی شود. اگر پدر و مادر و یا خواهر و برادر شما مبتلا به تالاسمی بوده و یا ناقل ژن تالاسمی هستند، بهتر است قبل از بچه دار شدن آزمایش های مربوط به تالاسمی را انجام دهید و با پزشک مشورت کنید. مشاور ژنتیک می تواند اطلاعاتی مانند میزان احتمال ابتلای فرزند شما به تالاسمی و میزان شدت آن را به شما بدهد.
معمولا تالاسمی خفیف باعث بروز هیچ علائمی نمی شود.
تالاسمی متوسط و شدید می توانند منجر به بروز علائم بیماری کم خونی شوند. به عنوان مثال ممکن است شما احساس ضعف، خستگی و تنگی نفس داشته باشید.سایر علائم این بیماری بستگی به شدن آن و نوع اختلالاتی که به وجود می آورد دارد.
کودکانی که تالاسمی شدید دارند ممکن است به کندی رشد کنند (عدم رشد)، استخوان بندی آنها شکل و ظاهری طبیعی نداشته باشد و همچنین اختلالاتی همچون مشکل در غذا خوردن، تب مکرر و اسهال داشته باشند.
پزشک در مورد سوابق پزشکی شما سوالاتی خواهد پرسید و سپس آزمایش هایی را برای شما تجویز خواهد کرد. ممکن است این آزمایش ها شامل موارد زیر باشند:
شمارش کامل سلول های خون (CBC)
آزمایش ژنتیک برای تشخیص ژن های مسبب تالاسمی
آزمایش میزان آهن خون
یک نوع آزمایش خون که به منظور تشخیص نوع تالاسمی، میزان انواع مختلف هموگلوبین خون را اندازه گیری می کنند.
اگر مبتلا به تالاسمی تشخیص داده شدید، سایر افراد خانواده ی شما نیز باید برای تشخیص تالاسمی به پزشک مراجعه کنند.
درمان بیماری به میزان شدت بیماری بستگی دارد.
بسیاری از مراکز پزشکی بزرگ و معتبر بخشی برای درمان اختلالات خونی دارند. این مراکز بهترین جا برای دریافت خدمات پزشکی می باشند.
تالاسمی خفیف نیازی به درمان ندارد. این نوع تالاسمی شایعترین نوع تالاسمی نیز می باشد.
تالاسمی متوسط ممکن است با فرایند انتقال خون و مکمل فولیک اسید درمان شود. فولیک اسید ویتامینی است که بدن شما برای ساختن سلول های قرمز خون به آن نیاز دارد.
در تالاسمی شدید ممکن است از روش های درمانی زیر استفاده شود:
انتقال خون
فولیک اسید
دارو هایی که برای شیمی درمانی از آنها استفاده می شوند (این دارو های به بدن کمک می کنند که هموگلوبین طبیعی تولید کند)
اگر عمل انتقال خون به صورت مکرر انجام شود ممکن است بدن شما با ازدیاد آهن مواجه شود. این حالت ممکن است به اعضای بدن شما و به ویژه به کبد شما آسیب وارد کند. از مصرف ویتامین هایی که حاوی آهن هستند اجتناب کنید همچنین از مصرف بیش از اندازه ی ویتامین C نیز بپرهیزید زیرا که این ویتامین به بدن کمک می کند که میزان آهن بیشتری را از مواد غذایی جذب کند. اگر شما بیش از حد مجاز آهن دریافت کرده باشید، پزشک شما ممکن است از کیلات درمانی (chelation therapy) استفاده کند. این روش درمانی نوعی دارو است که به حذف آهن های اضافه ی بدن شما کمک می کند.
درمانهایی که کمتر شایع هستند و برای تالاسمی های شدید استفاده می شوند عبارتند از:
پیوند خون و یا پیوند سلول های بنیادی مغز استخوان
برداشتن طحال به کمک عمل جراحی
مبتلایان به این بیماری باید هر سال واکسن آنفلوانزا دریافت کنند. در مورد دریافت واکسن پنوموکوک (Pneumococcal Vaccine) نیز با پزشکتان مشورت کنید. این واکسن می تواند از شما در مقابل عفونت های شدیدی که می توانند کم خونی شما را بدتر کنند و یا موجب بروز بیماری های شدیدی که از تالاسمی نشات می گیرند شوند، محافظت کند.
تالاسمی یک بیماری ارثی خونی است که باعث کم خونی خفیف یا وخیم می شود. این کم خونی به خاطر کاهش هموگلوبین و کمتر بودن گلوبول های قرمز خون از حد نرمال می باشد. هموگلوبین پروتئین موجود در گلوبول های قرمز خون است که اکسیژن را به همه ی قسمتهای بدن می رساند.
در افراد مبتلا به تالاسمی ژن هایی که هموگلوبین تولید می کنند وجود نداشته یا متغیر هستند (با ژن های معمولی متفاوت هستند). انواع وخیم تالاسمی معمولاً در اوایل کودکی تشخیص داده می شود و برای همه ی عمر در فرد باقی میمانند.
دو نوع اصلی تالاسمی، نوع آلفا و بتا می باشد و بنابر نام دو زنجیره ی پروتئینی که هموگلوبین نرمال می سازد نامگذاری شده اند. ژن های هر نوع تالاسمی از پدر و مادر به فرزندان منتقل می شود. تالاسمی آلفا و بتا، هر دو نوع وخیم و خفیف دارند.
تالاسمی آلفا زمانی اتفاق می افتد که یکی یا بیشتر از چهار ژن لازم برای ساخت زنجیره ی گلوبین آلفا از هموگلوبین وجود نداشته یا متغایر با نرمال باشد. کم خونی متوسط تا وخیم زمانی اتفاق می افتد که بیش از دو ژن متاثر باشند. شدیدترین نوع تالاسمی به تالاسمی آلفا ماژور شناخته شده است. این نوع تالاسمی ممکن است منجر به سقط جنین شود.
تالاسمی بتا زمانی اتفاق می افتد که یک یا هر دو ژن لازم برای ایجاد زنجیره ی گلوبین بتا هموگلوبین متغایر با نرمال باشد. شدت این بیماری به تعداد ژن های متاثر و ماهیت این نابهنجاری بستگی دارد. اگر هر دو ژن متاثر باشند، آنمی متوسط تا وخیم پدید خواهد آمد. نوع شدید تالاسمی بتا نیز به نام آنمی کولی معروف می باشد. آنمی کولی متداول ترین تالاسمی شدید در ایالات متحده می باشد.
تالاسمی های آلفا
تالاسمی آلفا “حامل خاموش”
تالاسمی آلفا خفیف، که تالاسمی آلفا مینور نیز نامیده می شود
بیماری هموگلوبین H
هیدروپس فِتال یا تالاسمی آلفا ماژور
تالاسمی به واسطه ی فقدان یا مغایرت ژن هایی است که در ساخت هموگلوبین موثر هستند. هموگلوبین پروتئین موجود در گلوبول های قرمز خون است که حامل اکسیژن هستند. افراد مبتلا به تالاسمی، نسبت به حد نرمال هموگلوبین و گلوبول های قرمز کمتری تولید می کنند. این مسئله به آنمی خفیف تا شدید منجر می شود.
ترکیب ممکن بسیاری از ژن های مغایر باعث ایجاد انواع مختلف تالاسمی می شود. تالاسمی همیشه موروثی است و از والدین به فرزندان منتقل می شود. افراد مبتلا به تالاسمی خفیف تا شدید، از هر دو والدین خود ژن هایی مغایر دریافت نموده اند. فردی که یک ژن یا ژن های تالاسمی را از یکی از والدین خود و از والد دیگر ژن نرمال دریافت نموده است، حامل به حساب می آید. در افراد حامل معمولاً، به جز کم خونی خفیف هیچ نشانه ای از بیماری دیده نمی شود، اما آنها نیز این ژن های مغایر را به فرزندان خود منتقل خواهند کرد.
هموگلوبین شامل دو نوع زنجیره ی پروتئین است که گلوبین آلفا و گلوبین بتا نامیده می شوند. اگر مشکل از جانب گلوبین آلفا باشد، این اختلال تالاسمی آلفا نامیده می شود. اما اگر مشکل از طرف گلوبین بتا باشد، تالاسمی بتا نام می گیرد. برای هر دو نوع این تالاسمی ها، هم نوع خفیف و هم شدید وجود دارد. تالاسمی بتا شدید معمولاً آنمی کولی نامیده می شود.
برای ساختن قسمت آلفای هموگلوبین، چهار ژن دخیل هستند—از هر والد دو ژن دریافت می شود. تالاسمی آلفا زمانی اتفاق می افتد که یک یا دو تا از این ژن ها وجود نداشته یا مغایر باشند.
افرادی که فقط یک ژن متاثر داشته باشند، حاملان خاموش نامیده میشوند و هیچ نشانه ی بیماری در آنها دیده نمی شود.
افرادی که دو ژن مغایر داشته باشند (که تالاسـمی آلفا مینور نامیده می شود) کم خونی خفیف داشته و حامل به شمار می روند.
افرادی که سه ژن متاثر داشته باشند آنمی متوسط تا شدید یا بیماری هموگلوبین H دارند.
نوزادانی که هر چهار ژن را مغایر دریافت می کنند (که تالاسـمی آلفا ماژور یا هیدروپس فِتال نامیده می شود) مدت کوتاهی پس از تولد جان می سپارند.
اگر دو حامل بیماری تالاسمـی آلفا صاحب فرزند شوند، نوزاد هم ممکن است به تالاسـمی خفیف یا شدید آلفا مبتلا شود و هم سالم باشد.
آنمی کولی به خاطر علائم و نشانه های آن از قبیل آنمی شدید، معمولاً در اوایل کودکی تشخیص داده می شود. در برخی از افرادی که مبتلا به انواع خفیف تری از تالاسمـی هستند، پس از یک آزمایش خون معمولی کم خونی تشخیص داده میشود. اگر کودکی مبتلا به کم خونی باشد، پزشکان به تالاسمی مشکوک خواهند شد و او را جزء افراد در خطر ابتلا به تالاسمی قرار خواهند داد.
برای تشخیص کم خونی که از کمبود آهن ایجاد می شود و کم خونی ایجاد شده توسط تالاسـمی، آزمایش سنجش میزان آهن خون انجام می گیرد. آنمی کمبود آهن به این علت اتفاق می افتد که بدن آهن کافی برای ساختن هموگلوبین ندارد. کم خونی در تالاسمی به خاطر کمبود آهن صورت نمی گیرد، و به خاطر مشکل زنجیره های گلوبین آلفا یا بتا است. مکمل های آهن هیچ تاثیری در بهبودی کم خونی ناشی از تالاسـمی ندارند، به خاطر اینکه کمبود آهن مشکل کار نیست.
افراد مبتلا به نوع متوسط بیماری (مثل تالاسـمی اینترمدیا) گهگاه به تزریق خون نیاز دارند، مثلاً زمانهایی که به خاطر یک بیماری دچار استرس می شوند. اگر بیماری فردی که مبتلا به تالاسمـی اینترمدیا است، وخیم تر شود و نیاز به تزریق های خون مکررتری پیدا کند، آن فرد دیگر تالاسمی اینترمدیا ندارد و به تالاسمی ماژور یا آنمی کولی مبتلا شده است.
افراد مبتلا به تالاسـمی های شدیدتر بیماری جدی و خطرناک دارند. این بیماران با تزریقات مکرر خون، درمان کی لیت آهن، و پیوند مغزاستخوان مورد درمان قرار میگیرند. اگر این افراد مورد درمان قرار نگیرند، در اوایل سنین کودکی جان خود را از دست می دهند. افرادی که درمان های خود را با موفقیت انجام دهند، حتی می توانند تا سی، چهل سالگی یا بیشتر زندگی کنند.
انواع شدید تالاسمـی با تزیرق خون مکرر درمان می شود. تزریق خون که با فرو کردن سوزن در رگ انجام می گیرد، خون حاوی گلوبول های قرمز نرمال را از طرف دهنده های سالم به بیمار می رساند. در درمان تالاسمی، تزریق خون روی برنامه و به طور منظم انجام می گیرد (هر ۲ یا ۴ هفته) تا میزان هموگلوبین و گلوبول های قرمز را در حد نرمال نگاه دارد. این درمان باعث می شود فرد مبتلا به تالاسمی بتواند فعالیت های عادی روزانه را به خوبی انجام دهد و عمری طولانی تر داشته باشد.
در درمان کی لیت آهن از داروهایی ویژه برای رفع آهن مازاد در افرادی که با تزریق خون زیاد با ازدیاد آهن روبه رو هستند، استفاده می شود. اگر این آهن اضافه برداشته نشود، ممکن است باعث آسیب رسیدن به اعضاء بدن مثل کبد، یا قلب شود.
این دارو، دیفروکسامین، زمانی بهتر عمل می کند که به آرامی زیر پوست وارد شود، که این کار معمولاً به وسیله ی یک پمپ سبک طی یک شب انجام می گیرد. این نوع درمان بسیار طاقت فرسا و سخت است و گاهی دردناک نیز می باشد، از اینرو افراد بسیاری پس از مدتی از انجام این درمان خودداری میکنند. یک قرص از درمان کی لیت آهن، دیفراسیروکس، در نوامبر ۲۰۰۵ برای استفاده در ایالات متحده تایید شد.
افرادی که دچار ازدیاد آهن هستند نباید از ویتامین ها و سایر مکمل هایی که حاوی آهن هستند استفاده کنند.
جراحی زمانی مورد استفاده قرار می گیرد که اعضاء بدن مثل طحال یا صفرا آسیب ببینند. برای مثال، اگر طحال ملتهب یا بزرگ شود، باید برداشته شود. اگر سنگ کیسه صفرا به وجود آید، صفرا نیز باید برداشته شود.
پیوند مغز استخوان یا سلول های بنیادین در کودکان بسیاری که مبتلا به تالاسـمی بوده اند، به طور موفقیت آمیزی انجام گرفته است. البته این عملیاتی بسیار خطرناک می باشد، اما برای آن دسته از کودکانی که واجد شرایط باشند، عمل می کند.
افرادی که مبتلا به تالاسـمی شدید هستند، بیشتر به بیماری هایی مبتلا می شوند که کم خونی آنها را بدتر هم می کند. به این افراد باید هر سال واکسن آنفولانزا و ذات الریه تزریق شود تا به این بیماری ها مبتلا نشوند.
اسید فولیک جزء ویتامین های B است که به ساخت گلوبول های قرمز خون کمک میکند. افراد مبتلا به تالاسـمی باید از مکمل های اسید فولیک استفاده کنند.
یک روز، این امکان مهیا خواهد شد تا تالاسـمی را در نوزاد هنوز متولد نشده با تزریق ژن نرمال به سلول های بنیادین نوزاد درمان کرد.
محققین در پی بررسی روش هایی برای بالا بردن تولید هموگلوبین جنینی در افراد مبتلا به تالاسمی هستند. هموگلوبین جنینی نوعی از هموگلوبین می باشد که قبل از تولد در بدن ساخته می شود. پس از تولد، بدن به جای تولید هموگلوبین جنینی، شروع به ساخت هموگلوبین بزرگسال می کند. بعضی از کودکان، یک ژن مغایر دارند که از این تغییر در ساخت هموگلوبین جلوگیری میکند و ادامه در ساخت هموگلوبین جنینی شدت بیماری آنها را کم می کند. محققین در پی راهی برای افزایش تولید هموگلوبین جنینی پس از تولد هستند.